

Pliage des protéines : Un module d'enseignement de l'I.A.
Introduction
Les protéines sont les piliers de la vie. Des catalyseurs biologiques aux neuromodulateurs, les interactions entre protéines pourraient représenter, dans le monde de l'IA, la boîte noire de la biologie moléculaire. Les étudiants en sciences apprennent souvent les quatre principales macromolécules - lipides, glucides, protéines et acides nucléiques - comme l'un des premiers sujets de leur cours de biologie. Cependant, seuls quelques étudiants acquièrent une compréhension approfondie de la manière dont ces molécules façonnent les organismes vivants. Les interactions moléculaires sont enseignées en chimie, alors que les molécules ne sont présentées que sous forme d'extraits dans la plupart des manuels de biologie (voir figure 1), ce qui empêche les étudiants d'établir des liens clairs entre les réactions chimiques apprises en chimie et le rôle critique des interactions moléculaires dans un contexte physiologique.
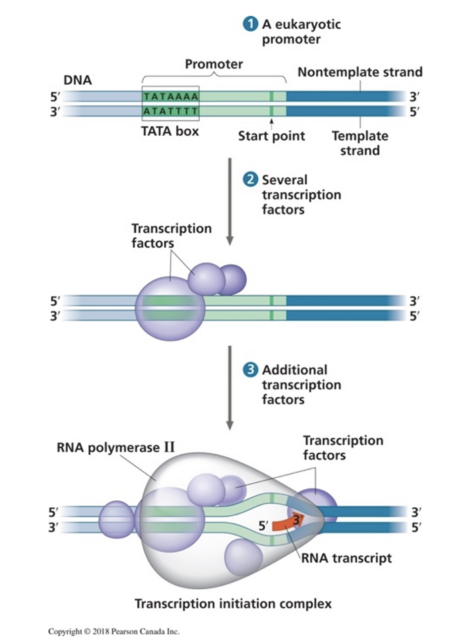
Figure 1. Représentation de l'ARN, des protéines et de l'enzyme (ARN polymérase), tirée d'un manuel de biologie populaire : Campbell Biology,3e édition canadienne.
Au cours des 70 dernières années, nous avons déchiffré comment les gènes et l'ADN codent pour des polypeptides spécifiques et nous pouvons prédire la chaîne d'acides aminés qui sera produite sur la base d'une séquence d'ADN. Cependant, notre capacité à prédire la structure tridimensionnelle d'un polypeptide à partir de ses composants de base était, jusqu'à récemment, très limitée. Les outils disponibles étaient lents et coûteux. La confirmation d'une structure tridimensionnelle nécessite souvent une cristallographie aux rayons X, la méthode utilisée par l'équipe de Rosalind Franklin pour générer la photo 51 qui a conduit à la découverte de la structure tridimensionnelle de l'ADN dans les années 1950.
Au cours des cinq dernières années, les méthodologies basées sur l'IA ont amélioré la vitesse et la précision de ces prédictions, ce qui a eu un impact considérable sur la recherche fondamentale et médicale.
Ce module pédagogique propose une introduction aux bases moléculaires de la production et du repliement des polypeptides, permettant de mieux comprendre l'impact crucial que les outils basés sur l'IA auront sur l'avenir des modèles de prédiction du repliement des protéines.
Module pédagogique
Tout d'abord, les concepts biochimiques fondamentaux relatifs à la production et à la structure des protéines seront explorés. Nous démontrerons ensuite la contribution de l'IA au domaine et présenterons des outils et des logiciels libres.
Le module est conçu comme trois activités indépendantes(parties 1, 2 et 3) qui peuvent être utilisées indépendamment ou comme une seule activité intégrée. Les parties I et II permettent aux étudiants de revoir/intégrer les concepts de la base moléculaire de la production et du repliement des protéines, et la partie III présente les outils et la contribution de l'IA, ainsi que des références/ressources/jeux supplémentaires pour approfondir cet aspect.
- Le module est conçu en tenant compte du fait que les étudiants ont des connaissances préalables sur la structure des protéines et les concepts de base de la synthèse des protéines cellulaires. En guise de préparation ou de révision, les sites web suivants sont de bonnes ressources pour les étudiants :
https://www.nature.com/scitable/topicpage/protein-structure-14122136/#
Première partie
- Le dogme central : revoir comment l'ADN code pour les protéines. En utilisant le code génétique (voir figure 2), les élèves appliqueront les concepts de codage de leur cours de programmation pour concevoir un algorithme dans lequel une série de codons d'ADN dans un gène calculera la séquence spécifique d'acides aminés dans une protéine (structure primaire). La collaboration entre les professeurs de biologie et les professeurs d'informatique est encouragée ici. Les élèves peuvent également réaliser l'activité sur un tableau blanc ou sur un document papier.
Dogme central

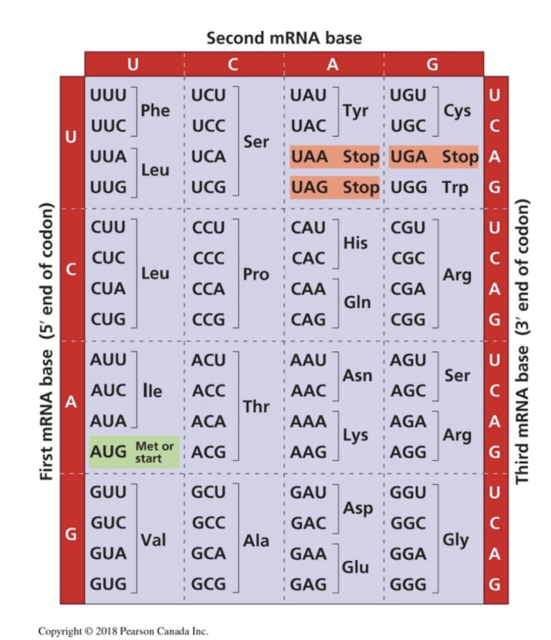
Figure 2. Le code génétique indique la correspondance entre chaque codon d'ARN (séquence de trois lettres) et un acide aminé spécifique, tel que déchiffré par Nirenberg en 1966. D'après Campbell,3e édition canadienne.
Pour s'aligner sur les activités suivantes, les élèves utiliseront le code ADN suivant, qui correspondra à la structure primaire de la protéine étudiée dans les deuxième et troisième parties de l'activité.
Instructions aux élèves
- En utilisant la séquence codante d'ADN suivante, 1- programmez ou 2- écrivez la séquence d'ARNm formée après la transcription. À partir de la séquence de l'ARNm, trouvez la structure primaire du polypeptide formé après la traduction. Pour simplifier, ne tenez pas compte des modifications de l'ARN telles que l'épissage.
Séquence de codage de l'ADN (peut être modifiée pour la séquence de modèles pour les étudiants plus avancés) :
5' ATG-CGT-TGG-CAA-GAA-ATG-GGT-TAT-ATT-TTT-TAT-CCT-AGA-AAA-TTA-CGT-TGA 3'
Transcription
ARNm :
Réponse : 5' AUG-CGU-UGG-CAA-GAA-AUG-GGU-UAU-AUU-UUU-UAU-CCU-AGA-AAA-UUA-CGU-UGA 3'
Traduction
Structure primaire du polypeptide :
Réponse : Met(M)-Arg(R)-Tryp(W)-Gln(Q)-Glu(E)- Met(M)-Gly(G)- Tyr(Y)-Ile(I)-Phe(F)- Tyr(Y)-Pro(P)- Arg(R)-Lys(K)-Leu(L)- Arg(R)
Partie II
- Sur la base de la polarité et des charges de chaque acide aminé (voir figure 3), les élèves appliqueront les concepts appris en chimie pour prédire les interactions entre les acides aminés. Les élèves doivent revoir les concepts d'hydrophobie, d'hydrophilie, de liaisons et, si possible, l'état d'énergie le plus bas d'une molécule.
L'article suivant peut intéresser les étudiants plus avancés :
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC21612/
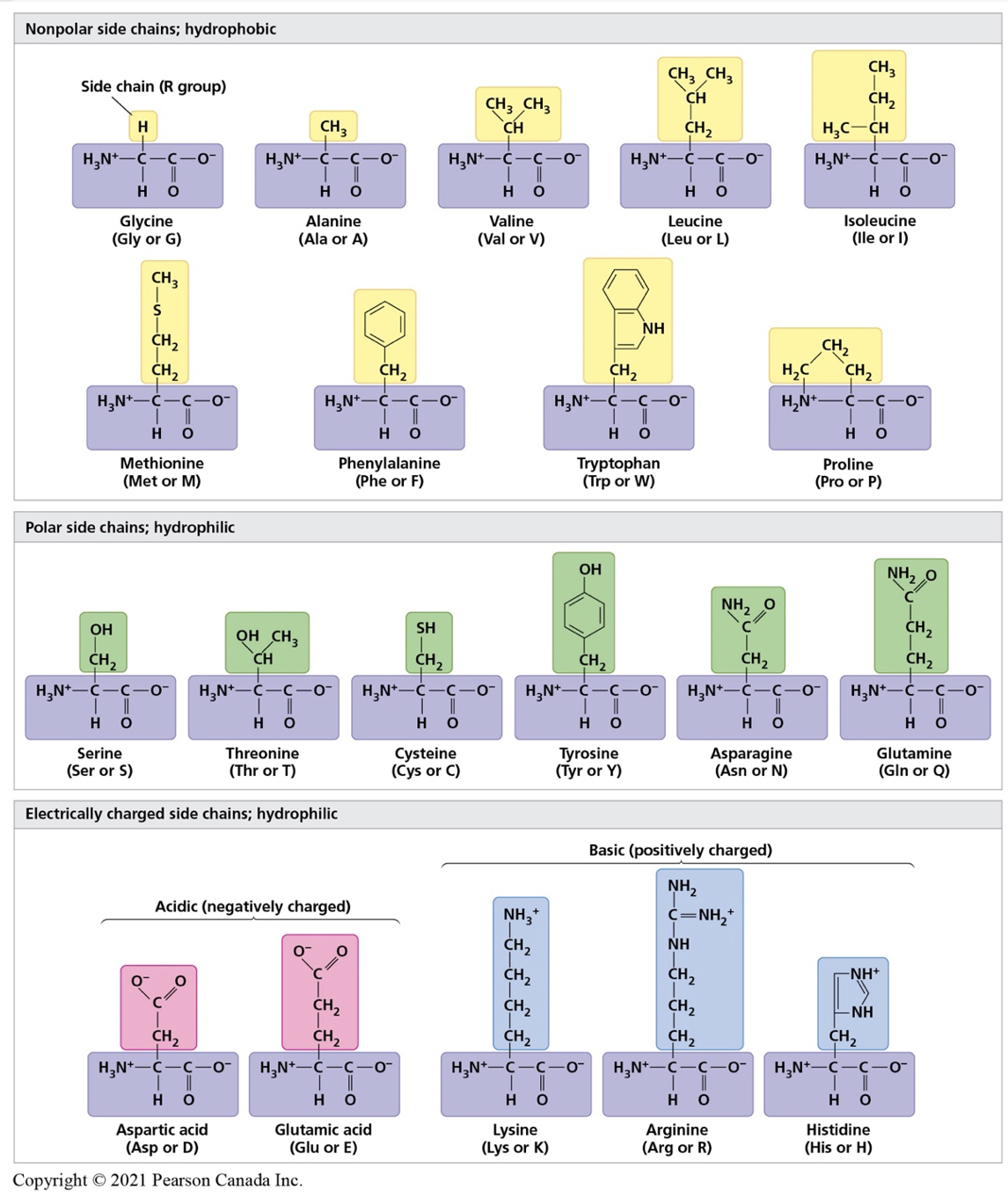
Figure 3. Propriétés des 20 acides aminés utilisés pour créer les protéines. Image tirée de la3e édition canadienne de Campbell.
Pour s'aligner sur les autres parties de l'activité, les élèves peuvent utiliser la séquence primaire de la protéine présentée pour prédire la structure tertiaire d'une protéine simple. L'enseignant doit imprimer l'acide aminé fourni, en noir et blanc, idéalement plastifié avec un petit trou de chaque côté de l'extrémité carboxyle et de l'extrémité amine, pour y attacher une ficelle.
Acides aminés à imprimer pour les élèves
Les enseignants doivent avoir à leur disposition des post-it de différentes couleurs (vert, jaune, rouge et bleu) ainsi que de la ficelle et des ciseaux.
Instructions aux élèves :
- Avant de commencer l'activité, discutez avec votre équipe des raisons pour lesquelles il est important pour la communauté scientifique de comprendre et de prédire la structure des protéines. L'article suivant peut alimenter votre discussion : https://www.nature.com/articles/s41586-021-03819-2
- Les acides aminés formant la structure primaire d'un polypeptide sont fournis (voir ci-dessous). Examinez le groupe R de chacun des acides aminés et indiquez la propriété chimique de chacun d'entre eux. Inscrivez vos résultats au crayon dans le tableau 1. Discutez avec votre équipe et votre professeur et confirmez vos réponses à l'aide de la figure 3.
-
- Indiquez la propriété correcte de chaque acide aminé fourni. Utilisez le code couleur ci-dessous et le post-it fourni pour identifier chaque acide aminé comme (à un pH physiologique de 7,4) :
- Polaire (vert), non polaire (jaune), chargé positivement (rouge) ou chargé négativement (bleu)
3. À l'aide du matériel fourni, attachez chaque acide aminé énuméré au suivant par des liaisons covalentes (des nœuds !). Les acides aminés doivent être attachés sur une ficelle dans l'ordre présenté, afin de représenter la structure primaire d'un polypeptide.
Structure primaire du polypeptide :
Met(M)-Arg(R)-Tryp(W)-Gln(Q)-Glu(E)- Met(M)-Gly(G)- Tyr(Y)-Ile(I)-Phe(F)- Tyr(Y)-Pro(P)- Arg(R)-Lys(K)-Leu(L)- Arg(R)
4. Pliez votre polypeptide en une éventuelle molécule de structure secondaire, puis tertiaire. Plier la chaîne (protéine) en une structure tertiaire qui respecterait les contraintes chimiques et physiques de base : les acides aminés chargés positivement et négativement s'attireront, les acides aminés non polaires seront situés vers l'intérieur de la protéine, etc. Plusieurs réponses sont possibles.
Pour une vue d'ensemble du repliement des protéines et de la dynamique de repliement des protéines, consultez les articles suivants :
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7545034/figure/F2/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7545034/
- https://bio.libretexts.org/Bookshelves/Biochemistry/Fundamentals_of_Biochemistry_(Jakubowski_and_Flatt)/01%3A_Unit_I-_Structure_and_Catalysis/04%3A_The_Three-Dimensional_Structure_of_Proteins/4.08%3A_Protein_Folding_and_Unfolding_(Denaturation)_-_Dynamics
-
- Pour chaque acide aminé de la structure primaire du polypeptide, noter la polarité/charge de chaque groupe R
Tableau 1. Polarité de divers acides aminés en fonction du groupe R.
| 1. Méthionine (M) : | 2. Arginine (R) : |
| 3. Tryptophane(W) : | 4. Glutamine(Q) |
| 5. Acide glutamique(E) : | 6. Glycine(G) : |
| 7. Tyrosine(Y) : | 8. Proline(P) : |
| 9. Lysine(K) : | 10. Leucine(L) : |
CLÉ DE RÉPONSE :
| 1. La méthionine : Non polaire | 2. Arginine : Chargée positivement (basique) |
| 3. Tryptophane : non polaire | 4. Glutamine : Polaire |
| 5. Acide glutamique : Chargé négativement (acide) | 6. Glycine : Non polaire |
| 7. Tyrosine : Polaire | 8. Proline : Non polaire |
| 9. Lysine : Chargée positivement (basique) | 10. Leucine : Non polaire |
Partie III
Le dernier thème présente les logiciels d'IA en biologie et la manière dont ils ont révolutionné le domaine de la prédiction de la structure des protéines.
Les premiers élèves peuvent jouer avec un jeu d'introduction au thème de l'évolution des protéines en utilisant l'alignement de séquences multiples, basé sur l'homologie de séquence et de structure.
Certains enseignants et étudiants pourraient être intéressés par la théorie qui sous-tend ces modèles, couramment utilisés dans la prédiction de la structure des protéines : https://www.sciencedirect.com/science/article/pii/B9780323897754000237
Modélisation de l'homologie :
https://blast.ncbi.nlm.nih.gov/Blast.cgi
https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/homology-modeling
Phylo :
"La comparaison des génomes de différentes espèces est l'une des techniques les plus fondamentales et les plus puissantes de la biologie moléculaire. Elle nous aide à déchiffrer notre ADN et à identifier de nouveaux gènes. Bien qu'il puisse sembler n'être qu'un jeu, Phylo est en fait un cadre permettant d'exploiter la puissance de calcul de l'humanité pour résoudre le problème de l'alignement de séquences multiples."
Ce jeu est développé par une équipe de McGill, et le programmeur peut ouvrir un "module de cours" adapté pour les enseignants intéressés.
Instructions aux élèves
- Dans cette activité, vous partirez de la structure primaire d'une protéine simple existante (voir les parties I et II) et utiliserez une base de données de protéines en libre accès pour explorer ce que les scientifiques savent de ce polypeptide.
-
- La base de données UniProt fournit la structure primaire et la structure tertiaire prédite de nombreux polypeptides. Accédez au site https://www.uniprot.org/
- Sélectionner les protéines Base de connaissances-UniProt
- Dans le champ de recherche situé en haut de la page, recherchez : MOTSC_HUMAN, puis cliquez sur le lien bleu qui apparaît sous Entry. Le numéro d'entrée est AOAOC5B5G6, le nom du gène est MT-RNR1 (voir le lien pour les étudiants avancés https://www.aging-us.com/article/102944/pdf ou https://pubmed.ncbi.nlm.nih.gov/25738459/ ).
- En utilisant les titres à gauche, familiarisez-vous avec votre protéine :
- Quel est le nom recommandé de la protéine ? ____________________________________
(réponse : Peptide dérivé de la mitochondrie MOTS-c)
-
- Où se trouve-t-il (localisation subcellulaire ou destination) ?
___________________________
___________________________
___________________________
(réponses : sécrété, mitochondrie, noyau)
2. Cliquez ensuite sur Structure et regardez la structure prévue.
Cliquez sur l'image générée pour zoomer et changer l'angle de vue. Double-cliquez sur un acide aminé pour afficher sa structure moléculaire. Comparez avec la structure prédite dans la partie II.
*Notez qu'il s'agit d'une protéine extrêmement courte, et que la structure tertiaire est donc plus simple et n'est pas représentative d'une protéine complexe dotée d'une structure secondaire et tertiaire sophistiquée. Pour simplifier l'exercice, revenez à la recherche et cherchez l'insuline humaine :
INS_HUMAN.
Félicitations, vous êtes maintenant familiarisé avec le monde du repliement des protéines et certains outils technologiques associés.
Il faut du temps et de la patience pour bien comprendre les nombreuses forces en jeu lorsqu'il s'agit du repliement des protéines, et même le Chat-GPT n'est pas en mesure de vous aider !
- Tu es maintenant prêt à explorer quelques jeux de pliage de protéines (attention, c'est addictif !).
Allez sur le site web de Fold it et téléchargez l'application. https://fold.it/. Vous pouvez maintenant explorer le côté éducatif ou passer directement à l'univers du joueur.
Vous pouvez également explorer le prédicteur de repliement des protéines qui a changé la donne : Le pliage Alpha de Deep Mind https://alphafold.ebi.ac.uk/
Fait intéressant : une organisation indépendante prévoit un "concours" semestriel pour fournir un mécanisme d'évaluation des méthodes de modélisation de la structure des protéines. Il est intéressant de voir le bond réalisé lorsque le nouvel outil d'Alpha Fold a été introduit. Consultez leur site web "Critical Assessment of Techniques for Protein Structure Prediction - CASP" (évaluation critique des techniques de prédiction de la structure des protéines - CASP) : https://predictioncenter.org/casp15/index.cgi
Références :
Campbell Biology,3e édition canadienne, 2021. Cain, M.L., Durnford, D.G., Minorsky, P.V., Moyes, C.D., Rawle, F.E., Reece, J.B., Scott, K., Urry, L.A., Wasserman, S

